HSCT or BMT accelerates brain atrophy rates and disability worsening in SPMS. #ClinicSpeak #MSBlog #MSResearch
I am revisiting this old paper to make it a Saturday trilogy of posts on HSCT.
HSCT (hemopoetic stem cell transplantation) or BMT (bone marrow transplantation) requires chemotherapy to ablate or wipe-out your immune system to allow the stem cell transplantation. The chemotherapy drugs that are used are neurotoxic, i.e. they damage the brain.
PwMS who already have pre-existing damage to their brain and spinal cords are particularly susceptible to the neurotoxic effects of chemotherapy. This is also driven by age; the older you are the worse you handle chemotherapy. The oncologists refer to this observation as chemobrain, which is particularly prevalent in the elderly.
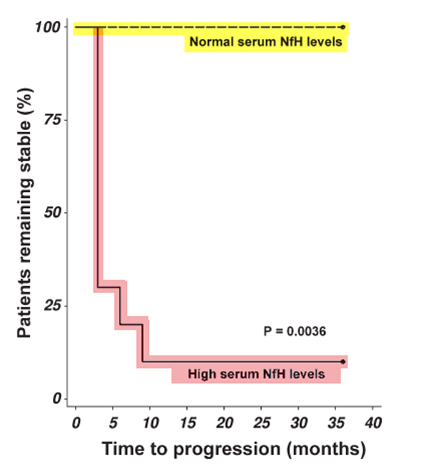
The following study below in which I was involved with shows that when pwMS are given chemotherapy they undergo increased neuronal loss, which is associated with worsening of their EDSS and greater brain atrophy. The data speaks for itself. The picture below is what we call a survival curve of EDSS worsening; you can see that the pwMS who had high blood levels of neurofilaments were much more likely to worsen than those who did not have raised neurofilament levels. Similarly, brain atrophy rates in pwMS were in the order of 2.1% per year in those who had a HSCT compared to only 1.2% per year in pwSPMS who did not have HSCT; the upper limit of normal for brain atrophy in healthy adults is generally accepted to be 0.4% per year. The bottom line is that if you have SPMS HSCT is likely to accelerate your disease worsening. As a result of these and similar observations most units have stopped doing HSCT in people with more advanced MS. However, with the advent of highly-effective DMTs such as alemtuzumab, natalizumab, fingolimod, daclizumab, ocrelizumab and cladribine the number of pwMS needing to be referred for HSCT should be small.

OBJECTIVE: Chronic neurotoxicity is a recognized long-term complication following chemotherapy in a range of diseases. Neurotoxicity adversely affects patients’ quality of life. The objective of this study is to examine whether there is evidence of acute neurotoxicity.
METHODS: This prospective study included MSers with secondary progressive multiple sclerosis (SPMS-BMT, n = 14) and hematological malignancies (HM-BMT, n = 17) receiving chemotherapy as preconditioning for bone marrow transplant. The control groups included SPMSers matched for demographic and clinical data (SPMS-PL, n = 14) and healthy controls (n = 14). Neurodegeneration was assessed at baseline and longitudinally (months 1, 2, 3, 6, 9, 12, 24, and 36), combining a clinical scale for disability (Expanded Disability Status Scale [EDSS]), a serum protein biomarker for neurodegeneration (neurofilaments, NfH-SMI35), and brain atrophy measures (magnetic resonance imaging).
RESULTS: Disability progression was significantly more acute and severe following chemotherapy compared to placebo. Immediately after starting chemotherapy, serum NfH-SMI35 levels increased in 79% (p < 0.0001) of SPMS-BMT MSers and 41% (p < 0.01) of HM-BMT patients compared to 0% of SPMS-PL MSers or healthy controls. In SPMS-BMT serum NfH-SMI35 levels were > 100-fold higher 1 month after chemotherapy (29.73ng/ml) compared to baseline (0.28ng/ml, p < 0.0001). High serum NfH-SMI35 levels persisting for at least 3 months were associated with sustained disability progression on the EDSS (p < 0.05). Brain atrophy rates increased acutely in SPMS-BMT (-2.09) compared to SPMS-PL (-1.18, p < 0.05).
INTERPRETATION: Neurotoxicity is an unwanted acute side effect of aggressive chemotherapy.
OBJECTIVE: Chronic neurotoxicity is a recognized long-term complication following chemotherapy in a range of diseases. Neurotoxicity adversely affects patients’ quality of life. The objective of this study is to examine whether there is evidence of acute neurotoxicity.
METHODS: This prospective study included MSers with secondary progressive multiple sclerosis (SPMS-BMT, n = 14) and hematological malignancies (HM-BMT, n = 17) receiving chemotherapy as preconditioning for bone marrow transplant. The control groups included SPMSers matched for demographic and clinical data (SPMS-PL, n = 14) and healthy controls (n = 14). Neurodegeneration was assessed at baseline and longitudinally (months 1, 2, 3, 6, 9, 12, 24, and 36), combining a clinical scale for disability (Expanded Disability Status Scale [EDSS]), a serum protein biomarker for neurodegeneration (neurofilaments, NfH-SMI35), and brain atrophy measures (magnetic resonance imaging).
RESULTS: Disability progression was significantly more acute and severe following chemotherapy compared to placebo. Immediately after starting chemotherapy, serum NfH-SMI35 levels increased in 79% (p < 0.0001) of SPMS-BMT MSers and 41% (p < 0.01) of HM-BMT patients compared to 0% of SPMS-PL MSers or healthy controls. In SPMS-BMT serum NfH-SMI35 levels were > 100-fold higher 1 month after chemotherapy (29.73ng/ml) compared to baseline (0.28ng/ml, p < 0.0001). High serum NfH-SMI35 levels persisting for at least 3 months were associated with sustained disability progression on the EDSS (p < 0.05). Brain atrophy rates increased acutely in SPMS-BMT (-2.09) compared to SPMS-PL (-1.18, p < 0.05).
INTERPRETATION: Neurotoxicity is an unwanted acute side effect of aggressive chemotherapy.
CoI: Prof. G was a co-author on this study.
I'm disappointed in this post. In HSCT there is an initial acceleration of brain atrophy (mostly pseudo atrophy, partly damage induced by the chemotherapy drugs). This continues on to year two, but after that brain atrophy returns to normal levels. Not 'normal for people with MS' levels. Actually normal levels of a little over 0.2% a year. You've seen the same HSCT followup studies I have.The REAL question is why Alemtuzumab shows an initial brain volume increase and thereon a less than normal brain volume decrease. Natalizumab shows an initial accelerated atrophy, theorised to be pseudoatrophy from a marked reduction in inflammation. Why does Alemtuzumab not show this? Is it not reducing inflammation as much? Is it magic? All I can theorise is that because the Alemtuzumab brain atrophy followup patients were treated much earlier in their course than the Natalizumab patients, that their brains had so much reserve they were immediately able to repair themselves enough once inflammation ceased to add to brain volume, but I doubt this can explain it.
Maybe profG wants to answer this one as he has been thinking about the atrophy data and has some ideas,,but best not steal his thunder….my first answer is that it goes to show what a useless outcome MRI brain atrophy is. Lamotrogine trial failed because of it. I was at an NMSS meetin yeg many years where brain volume was discredited, perhaps Grey matter volume change was better, yet role on ten years and some neurologists are still using it as a main outcome. May this has contributed to the failure to fin agents to treat progression. Heresy I know, but you have this array of data and how do you interpret it? We know that brain volume is subject to so many confounders, swelling, space filling with astrocytes and is a reflection of what has happened but not what is happening so the volume loss is not in sync with the damage yet people plough ahead with the same old same old, oblivious to the problem. We have had paper published showing cord area doesn't change but there is 60% nerve loss. Axonal loss in the multiple sclerosis spinal cord revisited.Petrova N, Carassiti D, Altmann DR, Baker D, Schmierer K.Brain Pathol. 2017 Apr 12. doi: 10.1111/bpa.12516.Likewise we know there is a relationship between nerve content and size.Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex.Carassiti D, Altmann DR, Petrova N, Pakkenberg B, Scaravilli F, Schmierer K.Neuropathol Appl Neurobiol. 2017 Apr 18. doi: 10.1111/nan.12405. [Epub ahead of print]Maybe is we can measure nerve loss in the blood using a biomarker this is how we should do the trials and forget the imaging.
Thanks for the response, that's extremely interesting. Just as I thought maybe we were getting somewhere with longer (5 year +, 10 year + in some cases) brain volume studies for different agents finally starting to come out. Neurofilament level testing does look really promising and I very much like the concept. As a marker specifically of neurodegeneration it's actually pretty awesome and I love the fact it can be carried over to any condition with a neurodegenerative component (vascular dementia, ALS, Alzheimers, etc). It should be incorporated into trials of new compounds immediately.I think brain atrophy testing still has a big place though, because from what I've read it does correlate quite well with disability (certainly much better than any measure like relapse rate, number of lesions on MRI etc).
Halcncod is right on the money. This study has a longer follow up time, and uses a stronger more toxic agent and result are in line with is coment:AbstractBackground: A cohort of patients with poor-prognosis multiple sclerosis (MS) underwent chemotherapy-based immune ablation followed by immune reconstitution with an autologous hematopoietic stemcell transplant (IA/aHSCT). This eliminated new focal inflammatory activity, but resulted in early accelerationof brain atrophy.Objective: We modeled the time course of whole-brain volume in 19 patients to identify the baselinepredictors of atrophy and to estimate the average rate of atrophy after IA/aHSCT.Methods: Percentage whole-brain volume changes were calculated between the baseline and follow-upmagnetic resonance imaging (MRI; mean duration: 5 years). A mixed-effects model was applied usingtwo predictors: total busulfan dose and baseline volume of T1-weighted white-matter lesions.Results: Treatment was followed by accelerated whole-brain volume loss averaging 3.3%. Both thebusulfan dose and the baseline lesion volume were significant predictors. The atrophy slowed progressivelyover approximately 2.5 years. There was no evidence that resolution of edema contributed tovolume loss. The mean rate of long-term atrophy was −0.23% per year, consistent with the rate expectedfrom normal aging.Conclusion: Following IA/aHSCT, MS patients showed accelerated whole-brain atrophy that was likelyassociated with treatment-related toxicity and degeneration of “committed” tissues. Atrophy eventuallyslowed to that expected from normal aging, suggesting that stopping inflammatory activity in MS canreduce secondary degeneration and atrophy.https://www.ncbi.nlm.nih.gov/pubmed/27246142
On the other hand This study with manly progressive patients shows another story:Patient characteristicsOf the 26 patients enrolled, 12 were female. Most had SPMS(n¼17) or primary progressive MS (n¼8) disease with a singlecase of RRMSThe estimated mean decrease in brain volume from baseline toþ2 years (change expressed as the mean of the differencesbetween baseline and 2 years; n¼12) was 2.6% (s.d.¼2.9%,P¼0.01). Nine of these patients also had MRI scans at þ5 years.The mean decrease in brain volume from þ2 to þ5 years was4.4% (s.d.¼5.9%, P¼0.06). Besides the sample size being small,these data do not suggest that the accelerated rate in brainvolume loss observed early after transplant decreases after 2 yearsin patients with advanced MS.Maybe Hsct is better for RRms https://www.ncbi.nlm.nih.gov/pubmed/22056644
"Axonal loss in the multiple sclerosis spinal cord revisited"Interesting study. Confirms that axonal damage in spinal cord is greater than demyelination. Also, demyelnation is greater in grey than white matter. The exact opposite of brain.More evidence that spinal MS is different than brain MS.
You casnt have more axonal loss than demyelination in the same place
Demyelination is greater in the grey than white matter in brain as well as spinal cord. Not sure about "brain MS being different from spinal MS" – they're both substrates of the same disease, probably playing out differently because of cellular/tissue composition/genetic background. Also note that focal demyelination apparently continues to drive axonal loss almost 30 years into the disease, suggesting that if we can stop this, there's a chance to alter the course of MS even at advanced stages.
Klaus, what do you think causes the diffuse degeneration in normal appearing white and grey matter that has never been subject to focal inflammation?
Dr Schmierer, please excuse my basic question: What do you mean by focal demyelination, what is this? Is there another kind of "systemic demyelination" or something?
focal demyelination means the demyelination is focused onto the lesion site the the alternative would be generalised systemic implies is is around the body so the peripheral nerve would be demyelinating
Dr KS, this is from 11 years ago:"Since there was little correlation between plaque load and axonal loss, the possibility that demyelination is not the primary determinant of spinal cord axonal loss warrants consideration."The contribution of demyelination to axonal loss in multiple sclerosis.https://www.ncbi.nlm.nih.gov/pubmed/16597651Apparently, there are strong arguments against your position that focal demyelination drives axonal damage.
"Dr KS, this is from 11 years ago"We think our paper (and further recent work by others) goes some way to rectify the misconception that axonal loss has essentially nothing to do with demyelination. Whilst in our study overall demyelination did not correlate with axonal loss (in line with the study you mentioned), once you look at lesions affecting specific pathways, and thoroughly exclude lesions affecting that pathway above and below the index lesion, it becomes quite evident that lesions lead to significant reduction in the number of axons. Check figures 3 & 7, and tables 5 & 6 in Natalia's paper.
Klaus, what do you think causes the diffuse degeneration in normal appearing white and grey matter that has never been subject to focal inflammation?Demyelination leads to forward (Wallerian) and backward (dying back) neuro/axonopathy; much of the diffuse change in non-lesional brain tissue is due to this process. Secondary damage through chronic microglia activation, gliosis, oxidative stress, mitochondrialdysfunction, among others, likely contributes to non-lesional changes in more advanced disease.
"..what do you think causes the diffuse degeneration in normal appearing white and grey matter ?"Spinal cord lesions took 20 years before they appeared in this patient. Likely long time build of lymphoid structureswhich then lead to spinal lesions in progressive ms.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5221476/https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5221476/figure/acn3377-fig-0001/"cortical pathology underlies progressive MS and that these changes are largely independent of underlying white matter lesions but rather might be associated with adjacent overlying meningeal lymphoid aggregates containing B cells and plasma cells.""In contrast to the cerebral hemispheres, the major white matter tracts in the spinal cord are located in a superficial location adjacent to the meninges where they might be susceptible to damage from effects of cytokines, free radicals, or autoantibodies. This anatomic feature raises the question of whether chronic meningeal inflammation might promote neocortical neurodegeneration in the underlying cerebral hemispheres, and demyelination in adjacent spinal cord white matter tracts."
I do quite like that hypothesis. The only issue is that demyelinating diseases like NMO do not have a secondary progressive phase despite being strongly demyelinating and inflammatory. What do you think separates MS from NMO in that regard?
"I do quite like that hypothesis. The only issue is that demyelinating diseases like NMO do not have a secondary progressive phase despite being strongly demyelinating and inflammatory. What do you think separates MS from NMO in that regard?"NMOSD is rather different from MS. It is an unequivocally autoimmune disease with – unlike MS – a defined antigen (aquaporin 4). A bout of NMOSD is more akin to a stroke in that all the damage, and the disability it causes, is done when a single lesion forms (or multiple thereof if a relapses occur). This basic concept remains true even if the spectrum of NMO “spectrum disorder” has been broadened by grouping disease phenotypes around a couple of specific antibody responses. An important cause of chronic deterioration in MS is the multiple-hit concept of long tract axons originally described by Kurtzke, more recently refreshed in our paper: http://ac.els-cdn.com/S2211034817300081/1-s2.0-S2211034817300081-main.pdf?_tid=93b3ffec-54d9-11e7-ba53-00000aacb361&acdnat=1497868092_4e496dc2e1155f5be02dedb9f491148a
Dr KS,There is an evident explanation for the contradicting findings about focal demyelination and axonal loss (total vs regional association): Both events are the result of some other damaging process. There is no causal relationship between demyelination and axonal loss. What is more, the damaging process leaves traces in these parts of the spine where you have found association between focal demyelination and axonal loss.I understand that this self-evident conclusion doesn't fit in any of the MS models you may accept, but I'm certain that the aforementioned parts are near the anchoring points of the denticulate ligaments. Can your co-autthors comment on that?
"What do you think separates MS from NMO in that regard?"MS and NMO seem to have alot of similarities including EBV.http://multiple-sclerosis-research.blogspot.com/2014/12/ebv-and-nmo.html
"I understand that this self-evident conclusion doesn't fit in any of the MS models you may accept, but I'm certain that the aforementioned parts are near the anchoring points of the denticulate ligaments."Not sure what you mean, perhaps let the cat out of bag and tell us? Do you believe MS is caused by mechanical stress via denticulate ligaments?There's plenty of evidence that lesions cause axonal damage/transection/loss.
"MS and NMO seem to have alot of similarities including EBV."Some similarities, but not a great deal. Here's a recent review I'd recommend: http://www.mayoclinicproceedings.org/article/S0025-6196(16)30849-7/pdf (open access).
Klaus, thank you for all the links and taking time out to answer questions. It is true that NMO is unequivocally autoimmune whereas MS is not. Progressive MS is the main MS puzzle."Demyelination leads to forward (Wallerian) and backward (dying back) neuro/axonopathy; much of the diffuse change in non-lesional brain tissue is due to this process. Secondary damage through chronic microglia activation, gliosis, oxidative stress, mitochondrialdysfunction, among others, likely contributes to non-lesional changes in more advanced disease."Why wouldn't this happen in NMO though? NMO is strongly demyelinating. It is true all the damage from NMO is caused when a lesion occurs, and slow burning damage does not happen. Why is this different with MS? What's different about the particular type of damage it is causing? Are there any conditions or circumstances wherein MS-like secondary progression occurs after well defined inflammatory damage? The brain has robusst mechanisms in place to repair itself, which is why progressive MS is such a puzzle. The brain does not deteriorate after a stroke because some process has been activated, it did not evolve like that.Why does it do so supposedly after inflammatory damage in MS?
"Do you believe MS is caused by mechanical stress via denticulate ligaments?"The idea is around 30 years now, but I haven't found any research that supports or disproves it. Personally, I'm convinced, though not by the arguments provided by Oppenheimer who proposed it in the first place. I wonder if you are aware of such a research, or whether you have a personal opinion about the possible correlation between the regional lesion load and the endpoints of denticulate ligaments. Maybe you plan to investigate it?I believe there are enough reasons for such an investigation, such as the the topology of MS lesions in the spine (sides & back), their unique shape (triangular as opposed to ovoid in brain) and the absence of a central vein, which in contrast is common in brain lesions.
And coming soon, the phrenology of MS 😉
Though I've always been very active and have had a lot of rough and tumble, I'm pretty sure I've had a lot less stress on my denticulate ligaments than the average rugby player or sumo wrestler. Any evidence MS is associated with heavy going contact sports? Thought not.
"The brain does not deteriorate after a stroke because some process has been activated, it did not evolve like that. Why does it do so supposedly after inflammatory damage in MS?"Remember there's disease activity in MS "between" clinical manifestations (relapses), some of which we can detect using MRI. This does generally not happen in stroke or NMO. If you add our length-dependency hypothesis, meaning multiple hits affecting the same tract systems in different places participate in driving disease deterioration makes more sense?
Thanks for the response Klaus. Are you saying that because the inflammatory process in MS hits more areas and is ongoing all the time (which is different to inflammation in NMO and obviously stroke) it has a higher chance of damaging the structure of the brain enough that it sets in process a secondary progressive, noninflammatory neurodegenerative process? I could definitely see that being the case.
"And coming soon, the phrenology of MS ;-)"What else are cancer treatments for MS? Pure lack of "phrena"
"And coming soon, the phrenology of MS ;-)"Correction, 39 years ago:https://www.ncbi.nlm.nih.gov/pubmed/683462The cervical cord in multiple sclerosis."It is argued that both these findings are explicable on the theory that mechanical stresses play a part in determining the site of lesions; that such stresses are commonly transmitted to the cord via the denticulate ligaments during flexion of the spine; and that many of the lesions are attributable to vascular leakages due to tension in the denticulate ligaments."
"Correction, 39 years ago: https://www.ncbi.nlm.nih.gov/pubmed/683462 The cervical cord in multiple sclerosis."Have you actually read that paper or just the abstract? Your claim spinal cord lesions wouldn't show veins is simply inaccurate, even based on this source – check figures 4-6.If you believe in a hypothesis, go an test it (or wonder why it hasn't been carried forward/confirmed/underpinned by others). Don't religiously refer to a single paper (by a single author, who could be a genius, but…) without taking note of the little bits of work that have been undertaken over the four decades since Oppenheimer's work was published.
"Have you actually read that paper or just the abstract?"I haven't read it. It costs 38$ to buy it. That's why i asked your opinion on this. Has it been disproved?"Your claim spinal cord lesions wouldn't show veins is simply inaccurate, even based on this source – check figures 4-6."I believe you. What about spinal lesion topology and shape?
You can download it here for one day:https://collect.qmul.ac.uk/down?t=R8RTNM0K1R0LA63K/4PUHB4CI84QDL6NNDM56KJG
Got it. Thank you VERY much.
The Oppenheimer paper is pure treasure. Picture 3 is shocking in correlating lateral lesions with denticulate ligaments binding points.Also, please note the analogies between Oppenheimer (a) and this (b):The denticulate ligament: anatomical properties, functionaland clinical significancehttps://link.springer.com/article/10.1007%2Fs00701-012-1361-x(a): "there is a preponderance of lesions in the lateral columns, abutting on the lateral surface, and having a roughly fan-shaped outline in transverse section."(b): "…each ligament had lateral triangular extensions oriented perpendicular to the long axis of the vertebral column."(a): "lesions in the cervical cord are about twice as frequent as those at lower levels."(b): "The collagenous fibers were larger in diameter and significantly more abundant at the cervical than the thoracic level." []"…at the cervical levels we observed that collagen fibers also extended from the DL to penetrate the spinal cord directly."
In practice what I have seen, since in Brazil the HSTC has been common in pwMS with a more aggressive course of the disease, is that those with SPMS who have done so tend to worsen yes, the degeneration even seems to increase. It seems that the inflammation "ends" but the neurodegeneration persists, and it seems to be even faster.
Just as a word of caution, the study posted by Prof G includes patients who had high dose radiation, myeloablative multi-agent chemotherapy, and allogeneic grafts. Given radiation is hugely neurotoxic in its own right, as is GVHD (which only happens in allogeneic transplsnts), and that the high intensity myeloablative chemo regimens are much more toxic… This isn't really comparable to the non radiation, low dose/single agent given in autologous HSCT for MS, as in most centres today.A little misleading from Prof G not to have made that clear, to be honest…Would he care to address this?
now now how is a neurologist to know the difference between the atrophy rates of different chemo drugs? it is far easier to paint everything with the same brush 🙂 Cinara, those with SPMS who have done so "tend to worsen"… which SPMS? People with SPMS who are EDSS 3.5 or people who are EDSS 6.5 and more?If it's the people with EDSS 6.5 and above as I suspect, then the problem (with increased atrophy rates) is not so much SPMS but the extent of existing damage.
Anon 5:16EDSS is a terrible proxy for extent of neurological damage. There are people with MS who have marked cognition issues, yet walk well.
"EDSS is a terrible proxy for extent of neurological damage. There are people with MS who have marked cognition issues, yet walk well."Yes, I know one person at least with over 40 lesions on the brain, terrible cognitive difficulties and walking fairly fine. I understand your point, but data is based on averages and my comments stands. I also refer you to early Burt et al HSCT studies on this issue (back when HSCT was being performed on SPMS and PPMS). Although numbers are small, look at the differences in progression free survival by EDSS . I get your point and I agree that it's not the best proxy marker, but it does the job for the purposes that I've mentioned above