Barts-MS rose-tinted-odometer: ★★★★★
I have been asked many times if we can cure someone who has MS. I have tried to explain what an MS cure may look like many times on this blog and have actually published articles defending the definition.
I explained in a previous post that you may be cured of your MS, but still, have worsening or progressive disease. The difference between progressive disease, which is due to previous MS damage and ageing is that the former should burn out, i.e. after a period of time, your worsening disability should eventually stop or flat-line. In comparison, MS-induced premature ageing is unlikely to stop. In comparison defining a cure in people who are young, with reserve capacity, who have been treated earlier is a much easier task.
From a biological perspective you can be cured but still have neurological deficits from previous damage, which need to be targeted with so-called ‘repair’ and ‘neuroregenerative’ therapies. These are separate processes and are independent of a so-called biological cure.
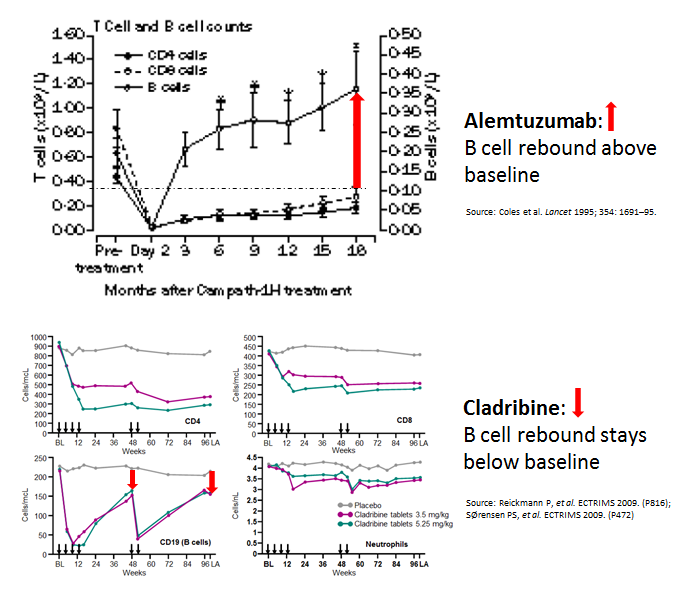
Based on our current understanding of MS a cure can only really occur in relation to IRTs (immune reconstitution therapies; e.g. alemtuzumab, cladribine & HSCT), i.e. treatments that are given as short courses that address the underlying ‘cause’ of MS. Maintenance treatments that need to be given continuously can’t cure MS, because when you stop the treatment MS disease activity tends to return and in some cases, particularly with anti-trafficking agents (natalizumab and fingolimod), to a greater extent than before, which we call MS rebound.
For arguments sake let’s say we have treated a group of pwMS early in the course of their disease with an IRT and they have gone into long-term remission with no evident disease activity (NEDA). How long should we wait before declaring a victory over their MS; 10, 15, 20 or 25 years? In the past, we have proposed defining a cure as NEDA at 15 years post-treatment as a starting point (see our MSARD Editorial below). Why 15 years? This is the most commonly accepted time-point used for defining benign MS and therefore it is a standard end-point that could potentially be accepted by the wider MS community. However, this may be wishful thinking many in the field are saying that we can’t cure MS, therefore, we should not even be having this discussion. Do you agree?
The average time to the onset of secondary progressive MS is ~14-15 years so one would expect to see a significant proportion of people manifesting with SPMS in this 15-year timeframe. If we have gotten the autoimmune hypothesis wrong and IRTs don’t work then I would estimate at least a third of treated subjects should have SPMS at 15 years. The problem with 15 years is that it is a long wait if you have MS. Many pwMS want to know ‘now’ if an IRT offers a cure, therefore we need data to convince the naysayers to support the ‘cure hypothesis’. Hopefully, convincing data, such as the HSCT data below, will change their minds and get them to at least offer IRTs to more of their patients.
In the past, I have proposed a deep phenotyping project to look at pwMS who are NEDA-2 post-IRT to see if we can find any evidence of ongoing inflammatory, or neurodegenerative, MS disease activity. I proposed interrogating them in detail and comparing them to a similar cohort of pwMS who are being treated with maintenance DMTs. Deep phenotyping is simply a term that refers to the interrogation of the CNS to see if the IRT has stopped ongoing damage and protected reserve capacity.
The study that has come closest to reaching this 15-year time point is the Canadian myeloablative HSCT cohort (see below). Mark Freedman, the principal investigator, has told me that all of these patients remain NEDA-2 (no relapses or MRI activity) although some have worsened in relation to their disability, which may be a result of previous damage and not ongoing MS disease activity. However, the most impressive observation is that this cohort of patients, who all had very active MS prior to HSCT, has ‘normalised’ their rate of brain volume loss or atrophy after an initial precipitous drop in brain volume due to pseudoatrophy and/or chemotherapy-induced neurotoxicity. Mark Freedman has also said that about a third of these patients, who have had lumbar punctures, have lost their OCBs (personal communication). However, the spinal fluid analyses have all been done quite early after HSCT hence we don’t know how many subjects who have reached 10 years of follow-up or more have persistent OCBs. Wouldn’t this be an interesting fact to know?
When the 10-year lumbar puncture and spinal fluid analysis was done in a group of Polish subjects treated with intravenous cladribine, 50% had lost their spinal fluid oligoclonal IgG bands (OCBs) at 10 years and this group of OCB-negative patients tended to have stable disease compared to those who hadn’t lost their OCBs. This is why we are doing the SIZOMUS (Ixazomib) and the DODO (high-dose ocrelizumab) studies to try and scrub the CNS clean of pathogenic B-cells and plasma cells that may be driving low-grade smouldering MS. Exciting? You bet! These two studies are one of the reasons I get up in the morning, look at myself in the mirror and say nobody can say Barts-MS isn’t doing innovative MS research.
The question I am now asking myself is switching a definition of a cure to a biological one a better strategy? This is a new line of thinking that has been brewing in my head for the last 12 months or so. If EBV is the cause of MS can we simply put pwMS into remission and clear them of EBV? This is why I want to do the iTeri and similar studies, i.e. to give an IRT and follow it with a drug that prevents EBV reactivation (antiviral) or scrubs B-cells of EBV (EBNA-1 antagonists).
I am sure many cynics will be saying no not Prof G thinking aloud. Yes, I am thinking aloud. If only a minority of pwMS treated with IRTs go into long-term remission why can we increase the proportion by using the induction-maintenance approach that targets the cause of MS? What do you think?
If you agree with this strategy I am going to need help to get the iTeri concept study funded.
DEFINING A CURE:
Banwell et al. Editors’ welcome and a working definition for a multiple sclerosis cure. Multiple Sclerosis and Related Disorders. 2013; 2(2):65-67.
…. Defining a cure in MS is a difficult task. How long should we wait before declaring a victory; 15, 20 or 25 years? Oncologists have back-tracked on this issue and instead of a cure they now prefer to use the term NEDD, or no evidence of detectable disease, at a specific time-point knowing full well that a limited number of subjects will relapse and present with recurrent disease after this point. We propose using the term NEDA, or no evident disease-activity, at 15 years as a starting point for defining a cure. Why 15 years? This is the most commonly accepted time-point used for defining benign MS and therefore it is a usual endpoint. In addition, the median time to the onset of secondary progressive MS is ~10-11 years (Kremenchutzky, Rice et al. 2006) and is well within the 15-year time window of our proposed definition of a cure. At present NEDA is defined using a composite of a) no relapses, or b) no EDSS progression, or c) no MRI activity (new or enlarging T2 lesions or no Gd-enhancing lesions) (Havrdova, Galetta et al. 2009; Giovannoni, Cook et al. 2011). This description is currently based on data that is routinely collected in contemporary clinical trials (Havrdova, Galetta et al. 2009; Giovannoni, Cook et al. 2011). The definition of NEDA will evolve with technological innovations and clinical practice, and in the future, it will almost certainly include MSer-related outcomes, grey matter disease activity, an index of brain atrophy and hopefully a CSF biomarker profile…..
References:
Giovannoni, G., S. Cook, et al. (2011). “Sustained disease-activity-free status in patients with relapsing-remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis.” Lancet Neurol 10(4): 329-337.
Havrdova, E., S. Galetta, et al. (2009). “Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study.” Lancet Neurol 8(3): 254-260
Kremenchutzky, M., G. P. Rice, et al. (2006). “The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease.” Brain 129(Pt 3): 584-594.
THE CURE #1?
Atkins et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet. 2016 Aug 6;388(10044):576-85.
BACKGROUND: Strong immunosuppression, including chemotherapy and immune-depleting antibodies followed by autologous haemopoietic stem-cell transplantation (aHSCT), has been used to treat patients with multiple sclerosis, improving control of relapsing disease. We addressed whether near-complete immunoablation followed by immune cell depleted aHSCT would result in long-term control of multiple sclerosis.
METHODS: We did this phase 2 single-arm trial at three hospitals in Canada. We enrolled patients with multiple sclerosis, aged 18-50 years with poor prognosis, ongoing disease activity, and an Expanded Disability Status Scale of 3.0-6.0. Autologous CD34 selected haemopoietic stem-cell grafts were collected after mobilisation with cyclophosphamide and filgrastim. Immunoablation with busulfan, cyclophosphamide, and rabbit anti-thymocyte globulin was followed by aHSCT. The primary outcome was multiple sclerosis activity-free survival (events were clinical relapse, appearance of a new or Gd-enhancing lesion on MRI, and sustained progression of Expanded Disability Status Scale score). This study was registered at ClinicalTrials.gov, NCT01099930.
FINDINGS: Between diagnosis and aHSCT, 24 patients had 167 clinical relapses over 140 patient-years with 188 Gd-enhancing lesions on 48 pre-aHSCT MRI scans. Median follow-up was 6.7 years (range 3.9-12.7). The primary outcome, multiple sclerosis activity-free survival at 3 years after transplantation was 69.6% (95% CI 46.6-84.2). With up to 13 years of follow-up after aHSCT, no relapses occurred and no GdGd-enhancing lesions or new T2 lesions were seen on 314 MRI sequential scans. The rate of brain atrophy decreased to that expected for healthy controls. One of 24 patients died of transplantation-related complications. 35% of patients had a sustained improvement in their Expanded Disability Status Scale score.
INTERPRETATION: We describe the first treatment to fully halt all detectable CNS inflammatory activity in patients with multiple sclerosis for a prolonged period in the absence of any ongoing disease-modifying drugs. Furthermore, many of the patients had substantial recovery of neurological function despite their disease’s aggressive nature.
THE CURE #2?
Rejdak et al. Cladribine induces long lasting oligoclonal bands disappearance in relapsing multiple sclerosis patients: 10-year observational study. Mult Scler Relat Disord. 2019 Jan;27:117-120.
Background: There has been long-term interest in cladribine as a drug for the treatment of MS. The current study focused on the effect of cladribine on oligoclonal bands (OCB) expression in the CSF in relapsing-remitting MS (RRMS) patients observed over 10 years.
Methods: 29 treatment-naive subjects with RRMS were prospectively enrolled and received induction therapy with subcutaneous parenteral cladribine (at a cumulative dose of 1.8 mg/kg; divided into 6 courses every 5 weeks given for 4-6 days, depending on patients’ body weight). Selected patients received maintenance doses in the follow-up period.
Results: Isoelectric focusing revealed that 55% of patients did not have OCB in CSF after cladribine treatment as compared to baseline testing when 100% of patients were positive for OCB. There were no significant differences in Expanded Disability Status Scale scores at baseline and at the end of treatment cycle between OCB-positive vs. OCB-negative subgroups. At the last follow-up, OCB-negative patients had lower disability compared to OCB-positive patients (p = 0.03).
Conclusion: Cladribine-induced immune reconstitution leads to long lasting suppression of intrathecal humoral response, which might be an additional mechanism that enhances the therapeutic effect on disease progression in RRMS patients.
Conflicts of Interest
Preventive Neurology
Twitter
LinkedIn
Medium
General Disclaimer: Please note that the opinions expressed here are those of Professor Giovannoni and do not necessarily reflect the positions of the Barts and The London School of Medicine and Dentistry nor Barts Health NHS Trust.