Barts-MS rose-tinted-odometer: ★★


Shortly after leaving Queen Square to take up my current position at Barts and The London Juliet Solomon, a good friend, and one of the research managers who had an office opposite me on the 6th floor of the Institute of Neurology sent me a signed copy of the ‘The Book of Regrets’ she had compiled as a present. She has asked celebrities to write essays on something they had regretted in their lives. The book has become a bestseller with all the proceeds of the sales going to support the National Hospital for Neurology and Neurosurgery. This is the kind of thing Juliet does; she has a very big heart. A mensch!

At the time I thought about what I would write if I was asked to contribute a chapter to the next edition of the book. I am still not sure, but from a professional perspective, my biggest regret is not being more proactive in derisking alemtuzumab as a treatment for MS. It has become clear to me that a small proportion of people with MS (pwMS) who are treated with alemtuzumab and HSCT early in the course of their disease are cured of MS when you use a contemporary definition of an MS cure.
If the infusion reactions, infections and secondary autoimmunity problems went away who would choose anything but alemtuzumab to treat their MS?
Infusion reactions: Can we reduce the infusion reactions of alemtuzumab? Yes, we can. Pre-treating with steroids starting the night before and moving from the intravenous to the subcutaneous route would make infusion reactions minor. So why hasn’t this been done? Money! MS centres/units make lucre out of infusing patients. I have used the subcutaneous route to avoid a second about of steroid-induced psychosis, to avoid steroid-induced metabolic mayhem in a patient with MS and type 1 diabetes and in a patient who developed avascular necrosis of one hip after his first course of alemtuzumab. It was remarkable; there were no major alemtuzumab infusion reactions despite these three patients being steroid-free. The pharmacodynamic data, i.e. the cell depletion data for the IV and SC routes are identical. The main reasons for us not switching all our patients to subcutaneous alemtuzumab was money, resource and inertia. In the recent past, we used to make money for the unit by giving infusions. Fortunately, with NHS block contracts, this perverse incentive has disappeared. Sanofi-Genzyme also provides contract nurses who come in to give the infusions and monitor the patients. If we converted our entire alemtuzumab administration programme to a subcutaneous route the workload would fall on our overworked nurses. Our nursing lead in our daycare unit reminded me of this. REGRET 1 we didn’t covert to sc alemtuzumab.
Secondary autoimmunity: What about preventing or reducing the incidence of secondary autoimmunity?
The immune system has many mechanisms in place to prevent autoimmunity. When you learn how the immune system works it is really quite surprising that autoimmunity is so uncommon. What the immunologists tell us is that there must be a series of underlying biological processes that are causing secondary autoimmunity and if we can work out what these are we can intervene and prevent this complication. This is what Joanne Jones and Alasdair Coles tried to do in Cambridge. Their hypothesis was that because the immune system reboots itself from peripheral memory cells it is more likely to result in an aberrant autoimmune response. They tried rebooting the immune system using more naïve cells from the thymus using the hormone palifermin, which stimulates the thymus to produce more naïve T-cells. Sadly it didn’t work, but at least they tried and they should be congratulated for doing this study.
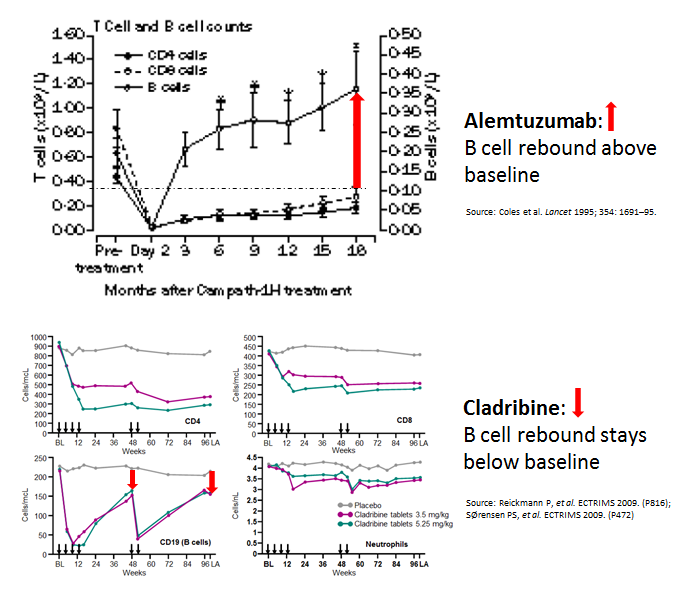
Interestingly, when you compare cladribine, another IRT, with alemtuzumab you can’t help but notice that the B-cell reconstitution profiles are very different. With alemtuzumab, they come back very quickly and overshoot their baseline values. We, and others, have hypothesised that if you change the profile of the B cell reconstitution with a small dose of the B cell depleting antibody rituximab you may be able to prevent this secondary autoimmunity. We are really talking about a very small dose of rituximab, i.e. 10-20mg, just enough to delay B cell reconstitution by 4-6 months. I proposed this concept to Genzyme 8 or 9 years ago, but without data to support the hypothesis they were not keen to support an exploratory study. Why didn’t I push to get this funded from another source? If we had done this study back then we would have had the results by now. Can you imagine how impactful this could be for pwMS if we could prevent secondary autoimmunity post-alemtuzumab? REGRET 2 no doing alemtuzumab-rituximab trial and not setting up an adaptive trial platform for testing multiple strategies to prevent secondary autoimmunity post-alemtuzumab.

Infections: One success story has been the derisking of alemtuzumab-associated infections with prophylactic antibiotics and antivirals, and the proactive approach to baseline infectious disease screening and vaccination. SUCCESS 1 reducing alemtuzumab-associated infections.
Anti-drug antibodies: Another success story has been exposing alemtuzumab’s problems with anti-drug antibodies (ADAs) and the development of an assay to screen for these antibodies. Why use a therapy, at great expense, that is not going to work because of neutralizing anti-drug antibodies. SUCCESS 2, anti-drug antibody screening.
A big problem that emerged was how alemtuzumab was licensed and used in the USA. The FDA has essentially licensed alemtuzumab with hand-cuffs, therefore, alemtuzumab was and is used as the DMT of last resort in the US. This led to it being used in an older more advanced cohort of pwMS who had comorbidities. In this group of patients, a new adverse event profile emerged, particularly vascular complications. This led to a safety review and the license of alemtuzumab’s use was changed and it is now only used infrequently, second or third -line and often in people with more advanced MS. I was personally involved with the original EMA submission; it was a real uphill battle to get alemtuzumab licensed as first-line therapy. Allowing the EMA to change how we use alemtuzumab, i.e. making pwMS have to wait to become eligible for the therapy is a travesty. We, Genzyme and MS community, should have made a more robust argument to the CHMP not to change alemtuzumab’s label. REGRET 3 not allowing alemtuzumab to be used first-line in active MS; it can only be used as a first-line agent in patients with rapidly evolving severe (RES) MS (two disabling attacks in a 12 month period). The problem is that very few patients have RES MS as they tend to be treated now before they have their second disabling relapse.
Finally, my colleagues. Apart from a small group of MSologists, and we know who we are, most MSologists don’t prescribe alemtuzumab. They find the therapy too difficult and risky to use. I have tried to educate and get more of my colleagues to at least offer alemtuzumab as an alternative to HSCT, but to no avail. In the UK, we were all geared up to do a head-2-head study of alemtuzumab vs. AHSCT. However, once ocrelizumab was licensed the MS community said they would not be able to recruit for this trial so it has now been converted into alemtuzumab or ocrelizumab vs. AHSCT trial. In reality, this study is going to be a head-2-head of ocrelizumab vs. AHSCT study. Not getting the wider MS community to understand how effective alemtuzumab is REGRET 4. Instead of success, we have a generation of refuseniks.
The question we need to ask ourselves is do we really want to throw the baby out with the bathwater? We have two, and possibly three, treatment strategies that may cure a minority of pwMS of having MS. Yes, CURE. However, alemtuzumab and HSCT are on the fringe of MS treatments. Why?
I suppose you are asking about the third option. It may be cladribine. The results of the ORACLE trial of cladribine in patients with clinically isolated syndromes (CIS) are quite remarkable. We are trying to recall the patients from the ORACLE study a decade or more later to see how many are still in remission and haven’t converted to MS. The problem we have is that cladribine is not even a treatment option for CIS despite this stunning data, hence we may be denying a large number of people with CIS, a relatively safe immune reconstitution therapy or IRT, that may prevent many of them from developing MS. The downside of this is the depressing fact that many MSologists still don’t treat CIS (see my blog post on watchful waiting).


Perumal et al. Subcutaneous administration of alemtuzumab in patients with highly active multiple sclerosis. Mult Scler. 2012 Aug;18(8):1197-9.
Alemtuzumab is an anti-CD52 monoclonal antibody with remarkable efficacy in relapsing multiple sclerosis (MS). In clinical trials and off-label use in MS, alemtuzumab has been administered intravenously (IV). Alemtuzumab is approved for chronic lymphoid leukemia as IV. Oncology guidelines recommend alemtuzumab subcutaneous (SC) over IV. There is no report of alemtuzumab SC in MS. We report two patients with highly active relapsing MS who were treated with SC alemtuzumab, had significant improvement and tolerated SC alemtuzumab well without the typical infusion-associated adverse events. SC alemtuzumab in MS warrants further studies as this may enhance patient convenience and minimize infusion-associated adverse events.
Leist et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014 Mar;13(3):257-67.
Background: Patients who develop relapsing-remitting multiple sclerosis (MS) present with a first clinical demyelinating event. In this double-blind, multicentre, randomised, phase 3 study we investigated the effect of oral cladribine on conversion to clinically definite MS in patients with a first clinical demyelinating event, when given at the same doses shown to be effective in relapsing-remitting MS.
Methods: Between Oct 21, 2008, and Oct 11, 2010, we recruited patients aged 18-55 years, inclusive, from 160 hospitals, private clinics, or treatment centres in 34 countries. Eligible patients had a first clinical demyelinating event within 75 days before screening, at least two clinically silent lesions of at least 3 mm on a T2-weighted brain MRI scan, and an Expanded Disability Status Scale score of 5.0 or lower. Patients with a first clinical demyelinating event ≤75 days before screening were randomly assigned (1:1:1) to receive cladribine tablets at cumulative doses of 5.25 mg/kg or 3.5 mg/kg or placebo. Randomisation was done with a central web-based randomisation system and was stratified by geographic region. Masking was maintained using a two-physician model. The primary endpoint of this 96-week study was time to conversion to clinically definite MS according to the Poser criteria. This study is registered with ClinicalTrials.gov, number NCT00725985.
Findings: Of 903 participants assessed for eligibility, 616 patients received cladribine 5.25 mg/kg (n=204), cladribine 3.5 mg/kg (n=206), or placebo (n=206). At trial termination on Oct 25, 2011, cladribine was associated with a risk reduction versus placebo for time to conversion to clinically definite MS (hazard ratio [HR] for 5.25 mg/kg=0.38, 95% CI 0.25-0.58, p<0.0001; HR for 3.5 mg/kg=0.33, 0.21-0.51, p<0.0001). Adverse events were reported in 165 (81%) patients in the cladribine 5.25 mg/kg group, 168 (82%) patients in the cladribine 3.5 mg/kg group, and 162 (79%) patients in the placebo group. We noted no increase in risk of adverse events with active treatment versus placebo apart from lymphopenia, which was a severe event in 10 (5%) patients in the 5.25 mg/kg group and four (2%) patients in the 3.5 mg/kg group.
Interpretation: Both doses of cladribine significantly delayed MS diagnosis compared with placebo. The safety profile of cladribine was similar to that noted in a trial in patients with relapsing-remitting MS. Further research could clarify the potential effects of oral cladribine treatment in the early stages of MS.
General Disclaimer: Please note that the opinions expressed here are those of Professor Giovannoni and do not necessarily reflect the positions of the Barts and The London School of Medicine and Dentistry nor Barts Health NHS Trust.


















