Barts-MS rose-tinted-odometer: ★★★★★ (rose-red; a climbing rose with thorns)


I have moved my treatment goal beyond NEIDA (no evident inflammatory disease activity) for my patients with MS. The new focus is on preventing end-organ damage. To achieve this we need to take off the blinkers that the Pharma industry has blinded us with. Our treatment target has to be smouldering MS, i.e. stopping disability progression, normalising brain volume loss, flattening neurofilament levels, stop slowly expanding lesions from getting bigger, clearing the CSF of oligoclonal bands and if possible promoting repair and recovery of the nervous system.
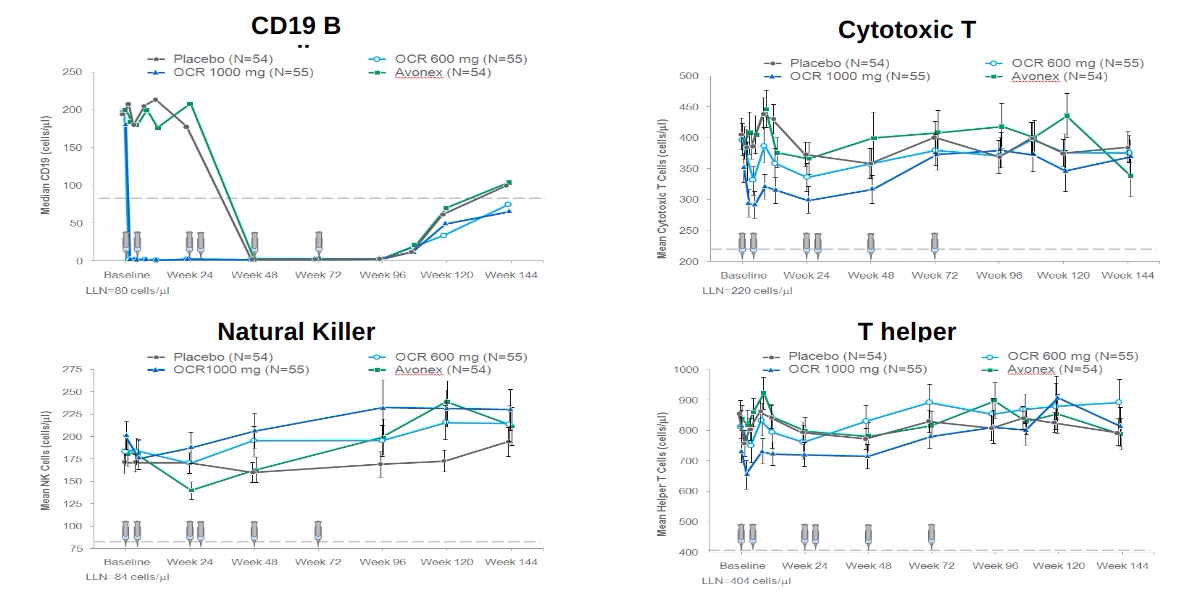
What good is to be free of relapses and focal MRI activity if you are getting worse? This is why the concept of using low dose anti-CD20 therapy is so flawed. It is clear that study subjects exposed to lower doses of ocrelizumab in the phase 3 trials did as well as those exposed to higher doses in relation to relapses and MRI activity, but not in relation to worsening disability (see slideshow below).

From this post-hoc analysis, it is clear that you need higher, and not lower, doses of anti-CD20 therapy at least initially as an induction strategy to purge the various B-cell compartments. We hypothesise these compartments house memory B-cells, which may be an important sanctuary for latent EBV and/or the highly autoreactive population of B-cells that drive and maintain the MS-state. This population of cells may reside in the deep tissues and/or the central nervous system. This is why we and others are testing CNS penetrant anti-B-cell strategies (ixazomib, cladribine, BTK inhibitors, etc.), i.e. we are going beyond the peripheral B-cell target.
However, I have hypothesized that once you have purged these compartments, say after 2 years of treatment you may not need to maintain such high doses of anti-CD20 therapy that will then suppress normal B-cell biology and immune responses, which result in long term complications. This is why I have proposed using ocrelizumab as an immune reconstitution therapy, i.e. high-dose upfront followed by no treatment and wait to see if MS remains in remission or disease-activity returns requiring additional courses. The latter is what we are proposing to do in the ADIOS study.
Even better would be two years of induction therapy with high-dose ocrelizumab followed by a maintenance therapy such as teriflunomide, leflunomide, IMU-838 (vidofludimus) or ASLAN003 (selective second-generation DHODH inhibitors), HAART (highly active antiretrovirals), famciclovir or another anti-EBV viral agent.
The hypothesis is to allow B-cell reconstitution after anti-CD20 therapy in the presence of an antiviral agent to prevent EBV reactivation and reinfection of new memory B cells. By doing this you will also be derisking the long-term immunosuppression associated with anti-CD20 therapies and prevent the development of hypogammaglobulinemia. This strategy will also allow patients to respond to vaccines.
However, if you want lower dose anti-CD20 therapy you will be able to start Ofatumumab very soon. Please remember ofatumumab was vastly superior to teriflunomide in suppressing relapses and MRI activity (Pharma’s blinkers) but was not superior to teriflunomide at slowing down brain volume loss in year two of the ASCLEPIOS I and II clinical trials (NCT02792218 and NCT02792231). Why?
The following is the fundamental question you should ask yourself.
So what would you choose your MS to be treated with; (1) low-dose anti-CD20, (2) high-dose anti-CD20, (3) high-dose anti-CD20 therapy followed by a maintenance treatment or (4) an immune-reconstitution therapy (cladribine, alemtuzumab or AHSCT)?
Sadly we can’t offer all of these choices to all of our patients with MS in the current NHS treatment landscape.

Hauser et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N Engl J Med. 2020 Aug 6;383(6):546-557.
Background: Ofatumumab, a subcutaneous anti-CD20 monoclonal antibody, selectively depletes B cells. Teriflunomide, an oral inhibitor of pyrimidine synthesis, reduces T-cell and B-cell activation. The relative effects of these two drugs in patients with multiple sclerosis are not known.
Methods: In two double-blind, double-dummy, phase 3 trials, we randomly assigned patients with relapsing multiple sclerosis to receive subcutaneous ofatumumab (20 mg every 4 weeks after 20-mg loading doses at days 1, 7, and 14) or oral teriflunomide (14 mg daily) for up to 30 months. The primary end point was the annualized relapse rate. Secondary end points included disability worsening confirmed at 3 months or 6 months, disability improvement confirmed at 6 months, the number of gadolinium-enhancing lesions per T1-weighted magnetic resonance imaging (MRI) scan, the annualized rate of new or enlarging lesions on T2-weighted MRI, serum neurofilament light chain levels at month 3, and change in brain volume.
Results: Overall, 946 patients were assigned to receive ofatumumab and 936 to receive teriflunomide; the median follow-up was 1.6 years. The annualized relapse rates in the ofatumumab and teriflunomide groups were 0.11 and 0.22, respectively, in trial 1 (difference, -0.11; 95% confidence interval [CI], -0.16 to -0.06; P<0.001) and 0.10 and 0.25 in trial 2 (difference, -0.15; 95% CI, -0.20 to -0.09; P<0.001). In the pooled trials, the percentage of patients with disability worsening confirmed at 3 months was 10.9% with ofatumumab and 15.0% with teriflunomide (hazard ratio, 0.66; P = 0.002); the percentage with disability worsening confirmed at 6 months was 8.1% and 12.0%, respectively (hazard ratio, 0.68; P = 0.01); and the percentage with disability improvement confirmed at 6 months was 11.0% and 8.1% (hazard ratio, 1.35; P = 0.09). The number of gadolinium-enhancing lesions per T1-weighted MRI scan, the annualized rate of lesions on T2-weighted MRI, and serum neurofilament light chain levels, but not the change in brain volume, were in the same direction as the primary end point. Injection-related reactions occurred in 20.2% in the ofatumumab group and in 15.0% in the teriflunomide group (placebo injections). Serious infections occurred in 2.5% and 1.8% of the patients in the respective groups.
Conclusions: Among patients with multiple sclerosis, ofatumumab was associated with lower annualized relapse rates than teriflunomide. (Funded by Novartis; ASCLEPIOS I and II ClinicalTrials.gov numbers, NCT02792218 and NCT02792231.).


General Disclaimer: Please note that the opinions expressed here are those of Professor Giovannoni and do not necessarily reflect the positions of the Barts and The London School of Medicine and Dentistry nor Barts Health NHS Trust.